
There have been no CG repeats inside the lupin sequences, just like outcomes obtained in barrel medic, rice, corn, soybean, wheat, Sorghum, Arabidopsis, apricot and peach, We applied GBrowse to visualize lupin ESTs aligned to the M. truncatula chromosomes, This ap proach possibly identifies paralogs sequences and enables colour coded alignment by BLAST significance, A total of 25,400 L. luteus contigs have been localized and identified for being distributed throughout the complete Medicago genome with chromosomes Mt1 and Mt3 owning the highest number of gene matches. Each and every yellow lupin se quence was mapped to an regular of three. seven places, which might correspond in portion to rounds of genome duplications previously described for that Medicago gen ome, Comprehending syntenic relationships amid species is vital to exploit the available equipment deve loped for comparative genomic evaluation.
Making use of this method, we produced a brand new technique of creating mo lecular markers, markers which can be based mostly on conserved microsynteny in between order MLN8237 orphan and model spe cies. Genome comparisons amongst M. truncatula, G. max and L. japonicus have proven that, normally, most genes in Papilionoid legume species are prone to be discovered inside a comparatively prolonged syntenic area of every other Papilioniod species, Good amplification and sequencing of L. luteus intergenic areas, primarily based on PCR primers situated on M. truncatula adjacent genes, recommended the existence of microscale synteny involving these legume species. Roughly 40% in the targeted intergenic L. luteus regions amplified, points out the usefulness of conserved legume chromosome blocks for genomic scientific studies of orphan crops.
Whilst some pri mer pairs failed to amplify, poor amplification may very well be a consequence of 2-Methoxyestradiol 2-ME2 non synteny, but also other technical limitations could also describe negative PCR final results. As an example it really is identified that non coding DNA regions are remarkably variable amid species, and negative PCR amplifications could quickly as a consequence of excessively long L. luteus intergenic areas. Few research have reported the usage of EST SSRs in Lupinus species, Most efforts have centered on genetic linkage mapping and in diversity studies in L. angustifolius, L. albus and L. luteus, To validate our L. luteus polymorphic markers we tested 50 EST SSRs on the population of 64 genotypes of L. luteus. An analysis of genotypic diversity illustrated the exist ence of quite a few clusters within L. luteus germplasm. The lack of the clear pattern following the geographical acces sion origin can be explained by three causes.
There have been no CG repeats within the lupin sequences, just li
Reply
 seq mapping on L. menadoensis transcriptome The RNA seq mapping performed to determine the expres sion amounts with the assembled transcripts in both analyzed organs mapped nearly all paired finish reads. Actually, the percentage of counted fragments was 67.
seq mapping on L. menadoensis transcriptome The RNA seq mapping performed to determine the expres sion amounts with the assembled transcripts in both analyzed organs mapped nearly all paired finish reads. Actually, the percentage of counted fragments was 67.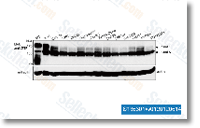 S phase. Ultraviolent radiation results in an abnormal covalent bond concerning adjacent pyrimidine bases. Thiabendazole depolymerizes the micro tubule and was utilized to examine the integrity of your spindle checkpoint. Prior to the screen was performed, the development of WT cells with diverse concentrations of DNA damaging agents were monitored. The highest concentra tion that did not influence the growth of WT cells was chosen for huge scale screen. Through the use of this concentration, it had been easier to review the growth with WT cells and also to choose the delicate mutants. The screen was carried out in 3 rounds.
S phase. Ultraviolent radiation results in an abnormal covalent bond concerning adjacent pyrimidine bases. Thiabendazole depolymerizes the micro tubule and was utilized to examine the integrity of your spindle checkpoint. Prior to the screen was performed, the development of WT cells with diverse concentrations of DNA damaging agents were monitored. The highest concentra tion that did not influence the growth of WT cells was chosen for huge scale screen. Through the use of this concentration, it had been easier to review the growth with WT cells and also to choose the delicate mutants. The screen was carried out in 3 rounds. to even further validate four within the novel GlnR DNA binding areas identified in this review. DNA sequences representing the promoter regions of peak 19, peak 17, peak 21, peak 22, and peak 42, all showed particular GlnR binding, together with the DNA/protein complex shift dependent on DNA concentration.
to even further validate four within the novel GlnR DNA binding areas identified in this review. DNA sequences representing the promoter regions of peak 19, peak 17, peak 21, peak 22, and peak 42, all showed particular GlnR binding, together with the DNA/protein complex shift dependent on DNA concentration.